Identification of substrates for Ser/Thr kinases using residue-based statistical pair potentials
Abstract
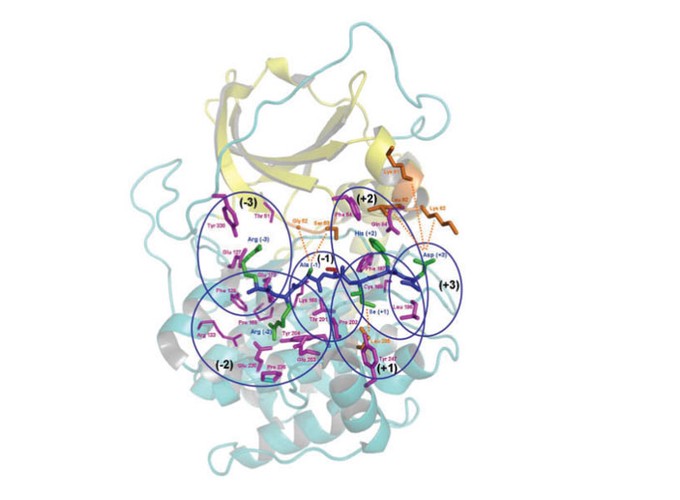
Motivation: In silico methods are being widely used for identifying substrates for various kinases and deciphering cell signaling networks. However, most of the available phosphorylation site prediction methods use motifs or profiles derived from a known data set of kinase substrates and hence, their applicability is limited to only those kinase families for which experimental substrate data is available. This prompted us to develop a novel multi-scale structure-based approach which does not require training using experimental substrate data. Results: In this work, for the first time, we have used residue-based statistical pair potentials for scoring the binding energy of various substrate peptides in complex with kinases. Extensive benchmarking on Phospho.ELM data set indicate that our method outperforms other structure-based methods and has a prediction accuracy comparable to available sequence-based methods. We also demonstrate that the rank of the true substrate can be further improved, if the high-scoring candidate substrates that are short-listed based on pair potential score, are modeled using all atom forcefield and MM/PBSA approach.